Epidermólisis Bullosa: la frágil piel de mariposa
Publicado el marzo 2, 2021 por Damary S. Jaramillo-Aguilar
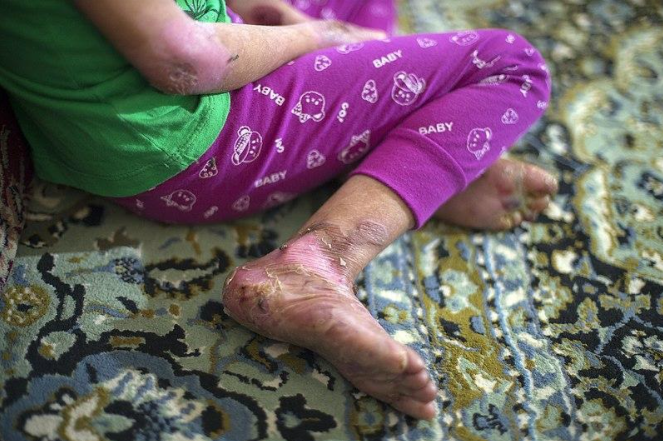
La epidermólisis bullosa (EB), conocida también como epidermólisis ampollosa, comprende un grupo de alteraciones genéticas –genodermatosis– que causan fragilidad de la piel y ampollas que usualmente son dolorosas, aun en caso de mínimos traumatismos; de ahí su denominación, piel de mariposa. [1, 2] También, la EB puede afectar a las mucosas de la vejiga, el estómago, y la boca. [3] En Estados Unidos, la prevalencia de EB es de 11.07 casos por cada millón de habitantes y su incidencia de 19.57 casos por cada millón de nacidos vivos. [2] No hay predilección por género o etnia. [2]
Las causas son, en general, hereditarias y pueden ser trastornos autosómicos dominantes o bien recesivos. Aunque también, muy poco vistos, suelen ser adquiridos y pueden aparecer a cualquier edad a consecuencia de trastornos autoinmunes, penfigoides e infecciones. [2]
Hasta ahora, son cuatro los tipos de EB conocidos:
- Epidermólisis ampollosa distrófica (EBD), trastorno hereditario –autosómico dominante (EBDD) o recesivo (EBDR)– causado por mutaciones en el gen COL7A1 que se encarga de codificar el colágeno tipo VII. Ante su deficiencia o síntesis defectuosa, se produce la aparición de ampollas subepidérmicas inmediatamente por debajo de la lámina densa, dando paso a la fragilidad mucocutánea, y en los casos más extremos, necrosis tras la evolución tórpida de las lesiones o su repetición. [4]
- Epidermólisis ampollosa simple (EBS), trastorno hereditario – autosómico dominante, pocos casos recesivos – causado por mutaciones en los genes K5 y K14 que codifican la producción de queratina y ciertas proteínas expresadas en los queratinocitos de la capa basal de la epidermis. Se caracteriza por ampollas intraepidérmicas y muy poca afectación sistémica, donde las lesiones disminuyen con la edad y suelen desaparecer sin dejar cicatrices. [5]
- Epidermólisis ampollosa tipo Kindler (SK), trastorno hereditario –autosómico recesivo –causado por una mutación en el gen que codifica la Kindlin– 1, un componente focal de contacto entre los queratinocitos basales. Son múltiples los planos de escisión, así: intradérmica, de unión o sublámina densa. [5] Las manifestaciones de la misma, suelen ir de leves a severas; así: leucoqueratosis labial, gingivitis, poiquilodermia, pseudosindactilia, estenosis esofágica, estenosis anal, colitis, fimosis severa, periodontitis, carcinoma agresivo de células escamosas, y otros. [6]
- Epidermólisis ampollosa juntural (EBJ), trastorno hereditario causado por una mutación en el gen LAMB3 que codifica la laminina-5, también en otros que codifican el colágeno tipo XVII y la integrina α6β4, de ahí que, haya separación de la lámina lúcida en la unión dermoepidérmica. Estos pacientes presentan afecciones de la mucosa oral, alopecia y anoniquia. Además, está asociado a anomalías congénitas del tracto genitourinario, muerte infantil/neonatal, y atresia pilórica congénita. [5]
El diagnóstico específico de la EB se hace mediante: 1) estudios histopatológicos, 2) análisis histoquímicos, 3) estudios moleculares de los genes de las proteínas implicadas y, 4) análisis prenatales, esto a familias con antecedentes de EB. [2]
Actualmente no existe cura para este trastorno; sin embargo, se dispone de una serie de medicamentos que ayudan a prevenir las infecciones asociadas y a reducir el malestar/inconformidad. Sumado a esto, se suele incluir: 1) cuidado adecuado de la piel, 2) tratamiento de infecciones adyacentes –generalmente por cándida albicans– y, 3) control de la alimentación. [3, 7] También, se suele intervenir quirúrgicamente al paciente, pero sólo en algunos casos, por ejemplo: 1) estenosis esofágica, 2) deformidades de la mano y, 3) carcinoma escamocelular. Además, vale mencionar que existen tratamientos que aún están bajo investigación, entre estos: terapia de proteínas y terapia genética. [7]

El pronóstico del paciente con EB dependerá de la severidad y progreso de la enfermedad. Así [8]:
- En pacientes con EBS, el pronóstico es excelente, con excepción de los pacientes con EBS generalizada, donde la mortalidad infantil temprana representa un riesgo.
- El pronóstico para EBJ severa, es sombrío, donde la mitad de los niños mueren antes de los 12 meses y, los jóvenes antes de los 25 años.
- En el caso de EBDD el pronóstico es muy similar al de la EBS, aunque, las ampollas y la cicatrización están presentes de por vida. Al contrario, en pacientes con EBDR, donde solo unos pocos pacientes mueren durante la infancia –generalmente por sepsis–, existe un riesgo acumulativo de por vida del 12% para muerte por insuficiencia renal crónica. Además, en edades avanzadas alrededor del 90% de pacientes habrá desarrollado carcinoma de células escamosas, y de estos el 87% habrán muerto.
- Para pacientes con SK el pronóstico es bueno.
La calidad de vida de la mayoría de pacientes con EB se ve afectada por los signos y síntomas propios de la enfermedad, además de, la falta de formación e información que se tiene sobre la misma. Los pacientes refieren pérdida de la autoestima y síntomas de dismorfia corporal, pérdida de la capacidad de afrontamiento, depresión, miedo intenso, ansiedad y aislamiento. En el ámbito social, la EB afecta la calidad de vida del paciente y, sobre todo, su entorno familiar, escolar, y laboral. Esto, a consecuencia del impacto económico que representa la misma, y el hecho de no poder realizar actividades cotidianas normales. [9]
En adición, según estudios realizados, en familias con niños afectados por EB es común observar: 1) la falta de interés y energía por parte de los padres al participar en actividades familiares y, 2) la alteración la dinámica de la pareja por la preocupación de tener otro niño con EB; lo que conduce a conflictos, como la separación/divorcio, que empeoran de cierta forma cuadro del paciente con EB. [10]
Finalmente, es de fundamental importancia el apoyo por parte de la familia y la sociedad a estos niños, jóvenes y adultos. Porque, recordemos, el apoyo y la permanencia también forman parte de un tratamiento eficiente.
Autora: Damary Silvana Jaramillo Aguilar – Universidad de Cuenca, Ecuador
Referencias:
- Genetic and Rare Diseases Information Center (GARD). Epidermolysis bullosa [Internet]. 2014 [cited 2020 Nov 23]. Available from: https://rarediseases.info.nih.gov/diseases/6359/epidermolysis-bullosa.
- Fine J-D. Epidemiology of Inherited Epidermolysis Bullosa Based on Incidence and Prevalence Estimates From the National Epidermolysis Bullosa Registry. JAMA Dermatol. 2016;152(11):1231-1238.
- National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS). Epidermolysis bullosa [Internet]. 2014 [cited 2020 Nov 23]. Available from: https://www.niams.nih.gov/health-topics/epidermolysis-bullosa.
- Shinkuma S. Dystrophic epidermolysis bullosa: a review. Clin Cosmet Investig Dermatol, 2015; 8: 275 – 284.
- Boeira V., et all. Inherited epidermolysis bullosa: clinical and therapeutic aspects. An Bras Dermatol, 2013 Mar – Apr; 88(2): 185 – 198.
- Pfendner EG, Lucky AW. Junctional Epidermolysis Bullosa [Internet]. 2008 Feb 22 [cited 2020 Nov 23]. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1125/.
- Clínica DAM, Especialidades Médicas. Epidermólisis ampollosa [Internet]. 2010 [cited 2020 Nov 23]. Available from: https://www.clinicadam.com/salud/5/001457.html.
- Epocrates. Epidermolysis bullosa, prognosis [Internet]. 2020 [cited 2020 Nov 23]. Available from: https://online.epocrates.com/diseases/74451/Epidermolysis-bullosa/Prognosis.
- Fine J., Johnson L., Winer M., Suchindran C. Impact of inherited epidermolysis bullosa on parental interpersonal relationship, marital status and family size. Br J Dermatol; 2005 May; 152(5): 1009 – 14. Citado, 23 de noviembre de 2020.
- Villar Á., et all. Abordaje interdisciplinar en el tratamiento de las heridas en epidermólisis bullosa. Enferm Dermatol; 2016; 10(29): 12 – 18. Citado, 23 de noviembre de 2020.